| Fibrodisplasia ossificante progressiva | |
|---|---|
 | |
| Specialità | reumatologia |
| Classificazione e risorse esterne (EN) | |
| OMIM | 135100 |
| eMedicine | 1112501 |
| Sinonimi | |
| Miosite ossificante congenita Malattia di Münchmeyer | |
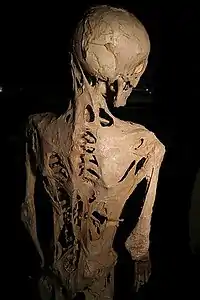
La fibrodisplasia ossificante progressiva (FOP), anche chiamata miosite ossificante congenita[1] o malattia di Münchmeyer, è una malattia genetica rarissima caratterizzata dalla presenza di focolai di ossificazione ectopici a livello del tessuto connettivo, in particolare tendini e legamenti, e del tessuto muscolare[2][3].
Epidemiologia
La prevalenza della fibrodisplasia ossificante progressiva è circa di un affetto ogni due milioni di persone[2]. Non presenta deviazioni di frequenza per etnia, sesso o popolazione geografica[4]. Nel mondo sono riportati circa 700 casi confermati, a fronte di una stima di circa 2500 persone affette[5]. La malattia viene diagnosticata nel primo decennio di vita, il 65% dei casi entro i quattro anni, e raramente dopo i vent'anni[2].
Eziologia
La malattia può avere trasmissione autosomica dominante, ma nel 90% dei casi è sporadica[2].
Clinica
L'esordio della malattia è caratterizzato dalla comparsa di tumefazioni che, col passare del tempo, assumono la consistenza del tessuto osseo, accompagnate da febbre e, talvolta, dolore. La localizzazione preferenziale è il collo, ma sono colpite anche spalle, dorso, bacino e radici degli arti[2]. La presenza di focolai di ossificazione nei muscoli ne limita progressivamente la funzione, soprattutto a livello dei muscoli respiratori della gabbia toracica; non vengono invece colpiti diaframma, lingua e sfinteri[3].
Trattamento
La terapia è sintomatica, con FANS e corticosteroidi utilizzati per trattare il dolore. L'utilizzo di bifosfonati per inibire la crescita ossea è controverso[2]. La rimozione delle aree di ossificazione, tramite intervento chirurgico, ha un altissimo tasso di recidiva[3]
Prognosi
La malattia è incurabile e progressiva, con gravi esiti invalidanti già intorno ai 15 anni[4], tali da rendere necessario l'uso di sedia a rotelle entro i 30 anni nella maggior parte dei pazienti[6]. Conseguentemente all'immobilizzazione forzata, il paziente muore per complicanze respiratorie, polmonite e asfissia[2], mediamente intorno ai 40 anni di vita[4].
Note
- ↑ Netter. Atlante di anatomia fisiopatologia e clinica. Apparato tegumentario, Bryan E. Anderson, Edra Masson, 2013, ISBN 8821437167
- 1 2 3 4 5 6 7 Canepa, pp. 2357-2363, 2002.
- 1 2 3 Netter, pp. 23, 2007.
- 1 2 3 FS. Kaplan, SA. Chakkalakal; EM. Shore, Fibrodysplasia ossificans progressiva: mechanisms and models of skeletal metamorphosis., in Dis Model Mech, vol. 5, n. 6, novembre 2012, pp. 756-62, DOI:10.1242/dmm.010280, PMID 23115204.
- ↑ Fopaction.co.uk Archiviato il 6 maggio 2012 in Internet Archive.
- ↑ R. Taslimi, S. Jafarpour; N. Hassanpour, FOP: still turning into stone., in Clin Rheumatol, novembre 2013, DOI:10.1007/s10067-013-2417-x, PMID 24253442.
Bibliografia
- Giuseppe Canepa, Trattato di ortopedia pediatrica, Padova, Piccin, 2002.
- Frank H Netter, Atlante di anatomia, fisiopatologia e clinica, vol. 8, Elsevier, 2007.
Voci correlate
Altri progetti
 Wikimedia Commons contiene immagini o altri file su fibrodisplasia ossificante progressiva
Wikimedia Commons contiene immagini o altri file su fibrodisplasia ossificante progressiva
Collegamenti esterni
- (EN) Fibrodisplasia ossificante progressiva, su Enciclopedia Britannica, Encyclopædia Britannica, Inc.
| Controllo di autorità | LCCN (EN) sh2006000680 · J9U (EN, HE) 987007542327505171 · NDL (EN, JA) 01187166 |
|---|