| Sindrome di Kasabach-Merritt | |
|---|---|
 | |
| Malattia rara | |
| Specialità | ematologia |
| Classificazione e risorse esterne (EN) | |
| ICD-9-CM | 287.39 |
| ICD-10 | D69.5 |
| OMIM | 141000 |
| MeSH | D059885 |
| eMedicine | 956136 |
| Sinonimi | |
| Emangioma con trombocitopenia | |
| Eponimi | |
| Haig Haigouni Kasabach Katharine Krom Merritt | |
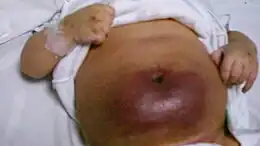
La sindrome di Kasabach-Merritt, conosciuta anche come emangioma con trombocitopenia[1], è una malattia rara, tipicamente infantile, caratterizzata dalla presenza di tumori vascolari congeniti che conducono a una riduzione del numero delle piastrine e, talvolta, ad altri disordini dell'emostasi[2] che possono essere pericolosi per la vita[3].
È stata descritta la prima volta dai pediatri Haig Haigouni Kasabach e Katharine Krom Merritt nel 1940[4].
Fisiopatologia
La sindrome è in genere causata da un emangioendotelioma o da un altro tumore vascolare, spesso presente alla nascita.[5][6] Anche se questi tumori sono relativamente comuni, è raro che causino il quadro clinico della sindrome di Kasabach-Merrit.
Quando questi tumori sono di grandi dimensioni o sono in rapida crescita, a volte possono sequestrare e consumare le piastrine, causando grave trombocitopenia. La combinazione di tumore vascolare e trombocitopenia definisce la sindrome. I tumori possono essere trovati nella estremità tronco, negli arti superiori e inferiori, nel retroperitoneo e nelle aree cervicali e facciali.[2]
Questo coagulopatia influisce anche su fattori di coagulazione, come il fibrinogeno che può peggiorare il sanguinamento. La coagulopatia può portare alla coagulazione intravascolare disseminata e persino alla morte.[2]
L'anemia emolitica secondaria alla distruzione microangiopatica (danno fisico) dei globuli rossi può essere espressa come lieve,.moderata o grave.[7]
Diagnosi
L'iter diagnostico per la condizione è diretto dai segni e sintomi di presentazione e può comprendere[7]:
- emocromo, studi di coagulazione e altre prove di laboratorio
- test di imaging biomedico (ecografia, TAC, risonanza magnetica, talvolta angiografia e più raramente esami di medicina nucleare)
- biopsia del tumore.
I pazienti mostrano in modo uniforme trombocitopenia grave, bassi livelli di fibrinogeno, elevati prodotti di degradazione della fibrina (a causa della fibrinolisi) e anemia emolitica microangiopatica.[2]
Trattamento
Il trattamento della sindrome di Kasabach-Merrit, in particolare nei casi più gravi, può essere complessa e richiede lo sforzo congiunto di più specialisti. Si tratta di una malattia rara, senza linee guida di trattamento o grandi studi randomizzati controllati che possano guidare la terapia.
Terapia di supporto
I paziente affetti dalla condizione possono risultare molto malati e quindi necessari di cure intensive. Essi sono a rischio di complicanze emorragiche tra cui emorragia intracranica. La trombocitopenia e la coagulopatia possono essere trattate con trasfusioni di piastrine e plasma, anche se è necessario prestare cautela a causa del rischio di sovraccarico di liquidi ed dell'insufficienza cardiaca da trasfusioni multiple. La possibilità di coagulazione intravascolare disseminata, una condizione pericolosa e difficile da gestire, è preoccupante. Farmaci anticoagulanti e antiaggreganti piastrinici possono essere utilizzati dopo un'attenta valutazione dei rischi e dei benefici.[7]
Il trattamento definitivo
In generale, il trattamento dei sottostanti tumori vascolari comporta la guarigione. Se la resezione chirurgica completa è fattibile, essa fornisce una buona opportunità per la cura (anche se può essere pericoloso operare un tumore vascolare in un paziente soggetto a sanguinamento).[7]
Se la chirurgia non è possibile, varie altre tecniche possono essere utilizzate per controllare il tumore:[2]
- embolizzazione (mediante radiologia interventistica) che può limitare l'apporto di sangue del tumore
- bendaggi compressivi esterni possono avere effetti simili
- alcuni farmaci, tra cui:
- corticosteroidi
- interferone-alfa
- chemioterapia (ad esempio vincristina)
- la radioterapia è stata usata, spesso con successo, ma ad oggi (2015) viene evitata quando possibile a causa del rischio di effetti negativi a lungo termine (ad esempio, il rischio di sviluppare un nuovo tumore in futuro).
Prognosi
La sindrome di Kasabach-Merrit ha un tasso di mortalità di circa il 30%. Per i pazienti che sopravvivono alla malattia acuta, la terapia di supporto può essere richiesta attraverso una graduale ripresa. Inoltre, i pazienti possono avere bisogno di cure da un dermatologo o chirurgo plastico per le residue lesioni estetiche. Sul lungo periodo di follow-up, la maggior parte dei pazienti presentano depigmentazione della pelle.[8]
Note
- ↑ James, William; Berger, Timothy; Elston, Dirk (2005). Andrews' Diseases of the Skin: Clinical Dermatology. (10th ed.). Saunders. ISBN 0-7216-2921-0.
- 1 2 3 4 5 Hall G, Kasabach–Merritt syndrome: pathogenesis and management, in Br J Haematol, vol. 112, n. 4, 2001, pp. 851–62, DOI:10.1046/j.1365-2141.2001.02453.x, PMID 11298580.
- ↑ Shim W, Hemangiomas of infancy complicated by thrombocytopenia, in Am J Surg, vol. 116, n. 6, 1968, pp. 896–906, DOI:10.1016/0002-9610(68)90462-5, PMID 4881491.
- ↑ Kasabach HH, Merritt KK, Capillary hemangioma with extensive purpura: report of a case, in Am J Dis Child, vol. 59, 1940, pp. 1063.
- ↑ Enjolras O, Wassef M, Mazoyer E, Frieden I, Rieu P, Drouet L, Taïeb A, Stalder J, Escande J, Infants with Kasabach–Merritt syndrome do not have "true" hemangiomas, in J Pediatr, vol. 130, n. 4, 1997, pp. 631–40, DOI:10.1016/S0022-3476(97)70249-X, PMID 9108863.
- ↑ el-Dessouky M, Azmy A, Raine P, Young D, Kasabach–Merritt syndrome, in J Pediatr Surg, vol. 23, n. 2, 1988, pp. 109–11, DOI:10.1016/S0022-3468(88)80135-0, PMID 3278084.
- 1 2 3 4 (EN) Medscape, Kasabach-Merritt Syndrome, su emedicine.medscape.com. URL consultato l'11 marzo 2016.
- ↑ Enjolras O, Mulliken J, Wassef M, Frieden I, Rieu P, Burrows P, Salhi A, Léauté-Labrèze C, Kozakewich H, Residual lesions after Kasabach–Merritt phenomenon in 41 patients, in J Am Acad Dermatol, vol. 42, 2 Pt 1, 2000, pp. 225–35, DOI:10.1016/S0190-9622(00)90130-0, PMID 10642677.
Bibliografia
- (EN) James, William; Berger, Timothy; Elston, Dirk (2005). Andrews' Diseases of the Skin: Clinical Dermatology. (10ª ed.). Saunders. ISBN 0-7216-2921-0
Altri progetti
 Wikimedia Commons contiene immagini o altri file su Sindrome di Kasabach-Merrit
Wikimedia Commons contiene immagini o altri file su Sindrome di Kasabach-Merrit